Semiconductor Theory Group
The Physics of Polymer Photonic Devices: Experiment and Theory

To model the optical response of a given system requires knowledge not only of the population of the system's excitations but also of the induced polarisations, as these are the sources for the propagating light field. To be able to meet these requirements, in our present project we use and develop a theoretical approach that combines the strength of (i) density-functional theory (DFT) calculations for molecular systems and (ii) a density-matrix theory based many-particle approach. The DFT calculations give detailed ab-initio insight into ground and excited vibronic state properties on the molecular level. This paves the way to formulate a microscopically founded model Hamiltonian describing a molecular solid-state system.
One key aspect in advancing polymer photonic and optoelectronic devices further, is the development of a profound theoretical understanding of the nonlinear optical excitation dynamics in organic semiconductor materials in the relevant excitation regimes. In describing and modelling inorganic semiconductor optoelectronic devices, such as amplifiers and lasers, equation of motion or density matrix theory approaches (the so-called Semiconductor Bloch Equations) have proven extremely successful. For organics systems, gaining a similarly high level of theoretical understanding as for their inorganic counterparts would be most desirable.
Self-trapping of the lowest singlet exciton in the centre of a fluorene dodecamer in the first excited state equilibrium geometry.
Great strides have been made over the last ten years in realising the enormous potential of organic semiconductor materials in that they are cheap, flexible and easily processed. The development of early polymer amplifiers and lasers today is following a similar trajectory to that of inorganic optoelectronic devices in the 1980's.
Typical organic materials of interest (e.g., polyfluorenes and fluorene based co-polymers) show optical properties that are, even in technologically relevant solid-state systems, essentially molecular in nature. Strong coupling of electronic and nuclear degrees of freedom in these systems gives rise to a vibronic substructure of molecular energy levels which is a fundamental ingredient in typical organics-based all-optical amplification and lasing schemes, and which is in strong contrast to inorganic semiconductor systems. On the other hand, important analogies with inorganic semiconductor optoelectronic systems exist, such as, e.g., the importance of excitonic effects, albeit with much higher binding energies, and the importance of exciton-exciton scattering processes at elevated excitation densities.
Starting from this Hamiltonian, density matrix equations of motion are derived to calculate (non-perturbatively in the external light field) the nolinear optical excitation dynamics of the system. Within this theoretical framework, important many-particle aspects such as inter-molecular excitation transfer and scattering, and relaxation and dephasing of vibronic excitations can systematically be included and studied. Also, direct inclusion of the propagation light field in the description allows to study important radiative effects such as amplified spontaneous emission (ASE) that is known to drastically influence device performance at high excitation densities.
Our research is carried out in close collaboration with Ifor Samuel at the University of St. Andrews, Organic Semiconductor Optoelectronics Group
Funding: Engineering and Physical Sciences Research Council (EPSRC).
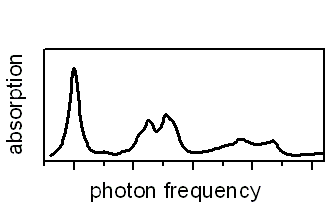
Low-temperature linear absorption of a fluorene octamer calculated from density-functional theory based ground and excited state information including electronic and vibrational degrees of freedom.
Prof. Ian Galbraith
Dr. Stefan Schumacher
Mr Jean-Christophe Denis
Mr Jack Wildman